Sickle Cell Disease: What You Need to Know Now
Sickle cell disease (SCD) is a genetic blood disorder that makes red blood cells stiff and crescent-shaped. That shape blocks blood flow, causes pain, and can damage organs over time. If you or someone you care for has SCD, there are clear steps that help reduce crises, avoid complications, and improve quality of life.
Recognize common problems fast
Pain episodes (called vaso-occlusive crises) are the most obvious sign — sudden, intense pain in the chest, back, arms, legs, or abdomen. Other frequent issues include chronic fatigue, shortness of breath, delayed growth in kids, and frequent infections. Watch for red flags that need urgent care: a fever above 101°F (38.3°C), sudden weakness or trouble speaking (possible stroke), chest pain and trouble breathing (acute chest syndrome), or prolonged painful erection (priapism). Don’t wait — these need ER evaluation.
Practical daily care and prevention
Hydration is simple and powerful: drink water often, especially during hot weather or exercise. Keep warm in cold weather and avoid extreme altitude when possible. Stay up to date on vaccines — pneumococcal and meningococcal vaccines lower infection risk. Take daily folic acid if your doctor recommends it to support red blood cell production. Avoid smoking and limit alcohol; both make complications more likely.
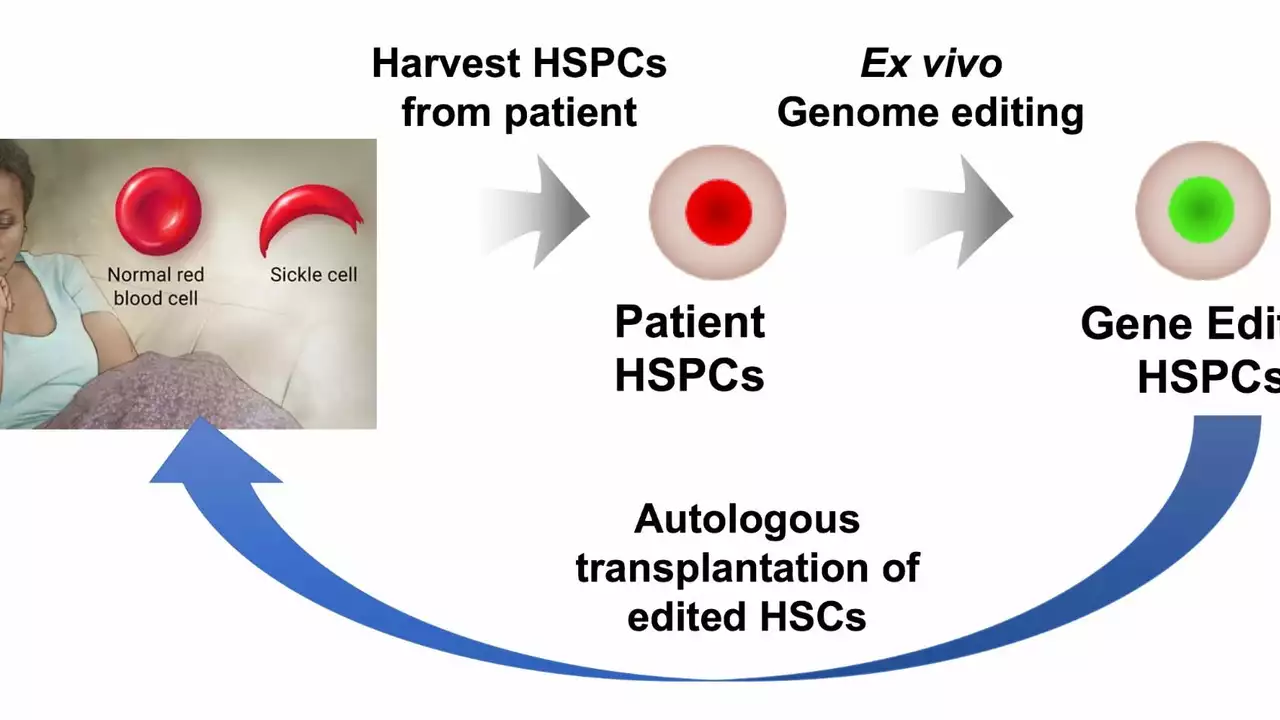
Medications that reduce crises: Hydroxyurea is the most common one. It raises fetal hemoglobin, lowers pain episodes, and decreases hospitalizations for many people. Some adults may benefit from newer drugs that lower sickling or inflammation — discuss options with a hematologist. Blood transfusions help in severe situations or to prevent stroke in children. A matched bone marrow (stem cell) transplant can cure SCD but has serious risks and isn’t an option for everyone.
Handle pain smartly: start pain relief early — use acetaminophen or NSAIDs for mild pain and prescription opioids for severe episodes under a doctor’s guidance. Use heat packs, gentle movement, and distraction techniques at home. If pain escalates despite home treatment, seek medical care: prolonged uncontrolled pain often needs IV medicines, fluids, and testing.
Plan ahead: create an emergency action plan with your hematologist that lists current meds, allergy info, baseline labs, and contact numbers. Wear medical ID that notes sickle cell disease. For families, newborn screening catches SCD early so care can start right away — that early start saves lives.
Think long term: regular checkups with a sickle cell center, eye exams, heart and kidney monitoring, and mental health support make a big difference. Genetic counseling helps people understand carrier status and family planning options. If you have questions about treatments or a new symptom, call your hematology team — quick action prevents many problems.
Sickle cell disease is manageable when you know what to watch for and follow a clear plan. Stay hydrated, keep up with vaccines and meds like hydroxyurea if recommended, and get urgent care for fever, chest pain, stroke signs, or severe uncontrolled pain.
I recently came across some intriguing research on Amiloride and its potential role in treating Sickle Cell Disease. Amiloride is a medication primarily used to treat high blood pressure and heart failure. However, studies have suggested that it could also help reduce painful symptoms and complications associated with Sickle Cell Disease. This is mainly due to its ability to inhibit certain ion channels, reducing the sickling of red blood cells. The prospect of Amiloride as a treatment for Sickle Cell Disease is exciting, and I'm eager to see how further research develops in this area.
Continue reading...